

Введіть код із SMS отриманого на номер
+38 (000) 000 00 00
Вибрати аптеку
Завантаження

 Каталог товарів
Каталог товарів
Вхід в особистий кабінет
Відновлення паролю
Підвердження входу
Відправити ще раз
Код надіслано.
Ви знову можете надіслати код підтвердження через 1:00
Реєстрація
Google
Аптека ВолиньФарм
- Дізнайтеся першими про нові акції та знижки
- Отримуйте % від суми покупки на бонусний рахунок
- Швидше оформляйте замовлення зі збереженою адресою та способом доставки
- Отримайте доступ до історії замовлень

 0
0
 0
0
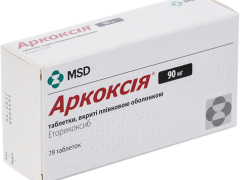
Аркоксія табл в/плівк обол 90мг блістер №28
Organon Central East Gmbh

Немає в наявності
Артикул:
6e98f5ce-0367-4b96-b4e9-26bf52a5314a
Атрибути
| Торгова назва | Organon Central East Gmbh |
| Країна виробництва | Швейцарія 2528 |
| Умови продажу | Рецепт |
| Діючі речовини | Еторикоксиб |
| АТХ-група | M01A H05 |
| Медикаменти | Нестероїдні протизапальні препарати |
Інструкція
Аркоксія (arcoxia) інструкція по застосуванню
Особливості застосування препарату
Спосіб застосування та дози Аркоксія
Склад
діюча речовина: etoricoxib;
- 1 таблетка містить 30 мг, 60 мг, 90 мг або 120 мг еторикоксибу;
допоміжні речовини: кальцію гідрофосфат безводний, целюлоза мікрокристалічна, натрію кроскармелоза, магнію стеарат;
оболонка таблетки: барвник Опадрай II синьо-зелений 39К11526 (для дозування 30 мг), Опадрай II зелений 39К11520 (для дозування 60 мг), Опадрай II білий 39К18305 (для дозування 90 мг), Опадрай II зелений 39К11529 (для дозування 120 мг), віск карнаубський;
склад барвника Опадрай II синьо-зелений 39К11526, Опадрай II зелений 39К11520, Опадрай II зелений 39К11529: лактоза, моногідрат; гіпромелоза; титану діоксид (Е 171); триацетин; індигокармін (Е 132); заліза оксид жовтий (Е 172);
склад барвника Опадрай II білий 39К18305: лактоза, моногідрат; гіпромелоза; титану діоксид (Е 171); триацетин.
Лікарська форма
Таблетки, вкриті плівковою оболонкою.
Основні фізико-хімічні властивості:
- Таблетки по 30 мг: двоопуклі таблетки яблукоподібної форми, вкриті плівковою оболонкою синьо-зеленого кольору, з гравіруванням <101> з одного боку та <ACX 30> з іншого.
- Таблетки по 60 мг: двоопуклі таблетки яблукоподібної форми, вкриті плівковою оболонкою темно-зеленого кольору, з гравіруванням <200> з одного боку та <ARCOXIA 60> з іншого.
- Таблетки по 90 мг: двоопуклі таблетки яблукоподібної форми, вкриті плівковою оболонкою білого кольору, з гравіруванням <202> з одного боку та <ARCOXIA 90> з іншого.
- Таблетки по 120 мг: двоопуклі таблетки яблукоподібної форми, вкриті плівковою оболонкою блідо-зеленого кольору, з гравіруванням <204> з одного боку та <ARCOXIA 120> з іншого.
Фармако-терапевтична група
Нестероїдні протизапальні та протиревматичні препарати. Коксиби. Код АТХ M01A H05.
Фармакологічні властивості
Фармакодинаміка.
Механізм дії
Еторикоксиб є пероральним селективним інгібітором циклооксигенази-2 (ЦОГ-2) у межах клінічного діапазону доз.
У ході клінічних фармакологічних досліджень лікарський засіб Аркоксія дозозалежно інгібував ЦОГ-2 без інгібування ЦОГ-1 при застосуванні у дозах до 150 мг на добу. Еторикоксиб не інгібує синтез простагландинів шлунка та не впливає на функцію тромбоцитів.
Циклооксигеназа відповідає за утворення простагландинів. Ідентифіковано дві ізоформи – ЦОГ-1 та ЦОГ-2. ЦОГ-2 є ізоформою ферменту, що індукується імпульсом прозапалення та розглядається як основний фактор, що відповідає за синтез простаноїдних медіаторів болю, запалення та гарячки. ЦОГ-2 також задіяна у процесах овуляції, імплантації та закриття артеріальної протоки, регуляції функції нирок та центральної нервової системи (індукція гарячки, відчуття болю, когнітивна функція). Також може брати участь у процесі загоєння виразок. ЦОГ-2 було ідентифіковано у тканині навколо виразки шлунка у людини, але значення для загоєння виразки не встановлено.
Ефективність
У пацієнтів з остеоартритом еторикоксиб у дозі 60 мг 1 раз на добу значно покращує стан при болях та оцінку пацієнта щодо стану захворювання. Ці позитивні ефекти спостерігалися вже на другий день лікування і зберігалися протягом періоду до 52 тижнів. У ході досліджень із застосуванням еторикоксибу в дозі 30 мг 1 раз на добу ефективність цього препарату перевищувала плацебо протягом 12 тижнів лікування (використовувалися оцінки, що застосовувалися в інших дослідженнях). Під час дослідження підбору дози еторикоксиб у дозі 60 мг демонстрував значно більш виражене покращення, ніж в дозі 30 мг, відносно усіх 3 основних кінцевих точок після 6 тижнів лікування. Застосування дози 30 мг при остеоартриті кисті не вивчалося.
У пацієнтів з ревматоїдним артритом еторикоксиб у дозі 60 мг та 90 мг 1 раз на добу значно покращував стан стосовно вираженості болю, запалення, а також рухливості. У ході досліджень з оцінки доз 60 мг та 90 мг позитивні ефекти зберігалися протягом 12-тижневого періоду лікування. У ході дослідження з оцінки дози 60 мг порівняно з дозою 90 мг, обидва дозування еторикоксибу – 60 мг 1 раз на добу та 90 мг 1 раз на добу – були більш ефективні ніж плацебо. Доза 90 мг була ефективніша, ніж доза 60 мг відповідно до методу Загальної оцінки болю пацієнтів (0–100 мм візуальна аналогова шкала), з середнім покращенням -2,71 мм (95 % ДІ:
-4,98 мм, -0,45 мм).
У пацієнтів з нападами гострого подагричного артриту еторикоксиб у дозі 120 мг 1 раз на добу протягом 8 днів полегшував біль у суглобах середнього та тяжкого ступеня та запалення порівняно з індометацином у дозі 50 мг 3 рази на добу. Зменшення вираженості болю спостерігається вже через 4 години після початку лікування.
У пацієнтів із анкілозуючим спондилітом еторикоксиб у дозі 90 мг 1 раз на добу забезпечує значне покращення при болю у хребті, запаленні, обмеженні рухів, а також покращує функціональну здатність. Клінічні переваги еторикоксибу спостерігалися на другий день після початку терапії і зберігалися упродовж 52-тижневого періоду лікування. У ході другого дослідження з оцінки дози 60 мг порівняно з дозою 90 мг, еторикоксиб у дозі 60 мг 1 раз на добу та 90 мг 1 раз на добу продемонстрував схожу ефективність порівняно з напроксеном 1000 мг щоденно. У пацієнтів, що не демонстрували адекватної відповіді під час застосування дози 60 мг щоденно протягом 6 тижнів, підвищення дози до 90 мг щоденно покращувало оцінку інтенсивності болю у спині (0–100 мм візуальна аналогова шкала) порівняно з продовженням прийому 60 мг щоденно, з середнім покращенням -2,70 мм (95 % ДІ: -4,88 мм, -0,52 мм).
Під час клінічного дослідження післяопераційного зубного болю еторикоксиб у дозі 90 мг застосовувався 1 раз на добу до трьох днів. У підгрупі пацієнтів з помірним болем в початковому стані еторикоксиб у дозі 90 мг демонстрував знеболюючий ефект, подібний до такого в ібупрофену 600 мг (16,11 проти 16,39; P=0,722), і перевищував ефект парацетамолу/кодеїну 600 мг/60 мг (11,00; P<0,001) і плацебо (6,84; P<0,001), що визначалося за показником повного полегшення болю через 6 годин (TOPAR6). Кількість пацієнтів, які повідомляли про застосування препаратів екстреного знеболення протягом 24 годин, склала 40,8 % у групі застосування еторикоксибу 90 мг, 25,5 % у групі застосування ібупрофену 600 мг кожні 6 годин і 46,7 % у групі застосування парацетамолу/кодеїну 600 мг/60 мг кожні 6 годин порівняно з 76,2 % пацієнтів, які приймали плацебо. У цьому дослідженні початок аналгезивної дії (відчутне полегшення болю) 90 мг еторикоксибу спостерігалося вже через 28 хвилин після прийому препарату.
Безпека
Міжнародна дослідницька програма тривалого застосування еторикоксибу і диклофенаку при артриті (MEDAL)
Програма MEDAL була проспективно розробленою програмою відносно результатів щодо безпеки з боку серцево-судинної системи, отриманих за об’єднаними даними трьох рандомізованих, подвійно сліпих, контрольованих активним препаратом порівняння досліджень (дослідження MEDAL, EDGE II і EDGE).
У дослідженні MEDAL, що було направлене на визначення впливу на серцево-судинну систему, брали участь 17804 пацієнти з остеоартритом (ОА) і 5700 ‒ з ревматоїдним артритом (РА), які застосовували еторикоксиб у дозі 60 мг (ОА) чи 90 мг (ОА і РА) або диклофенак у дозі 150 мг на добу протягом в середньому 20,3 місяця (максимально ‒ 42,3 місяця, медіана ‒ 21,3 місяця). У цьому дослідженні були зафіксовані тільки серйозні побічні реакції і припинення прийому препарату внаслідок виникнення будь-яких побічних реакцій.
У ході досліджень EDGE і EDGE II порівнювали шлунково-кишкову переносимість еторикоксибу і диклофенаку. У дослідженні EDGE брали участь 7111 пацієнтів з ОА, які отримували еторикоксиб у дозі 90 мг на добу (у 1,5 раза вище рекомендованої дози для лікування ОА), або диклофенак у дозі 150 мг на добу протягом в середньому 9,1 місяця (максимум ‒ 16,6 місяця, медіана ‒ 11,4 місяця). У дослідженні EDGE II брали участь 4086 пацієнтів з РА, які отримували лікування еторикоксибом у дозі 90 мг на добу або диклофенаком у дозі 150 мг на добу протягом в середньому 19,2 місяця (максимум ‒ 33,1 місяця, медіана ‒ 24 місяці).
У об’єднаній програмі MEDAL брали участь 34701 пацієнт з ОА і РА, які отримували лікування упродовж періоду в середньому 17,9 місяця (максимум ‒ 42,3 місяця, медіана ‒ 16,3 місяця); приблизно 12800 пацієнтів отримували лікування понад 24 місяці. У пацієнтів, зареєстрованих у цій програмі, були різні початкові чинники ризику стосовно серцево-судинної системи і шлунково-кишкового тракту (ШКТ). Пацієнти з нещодавно перенесеним інфарктом міокарда, аортокоронарним шунтуванням або черезшкірною коронарною ангіопластикою упродовж 6 місяців до реєстрації в дослідженні були виключені з дослідження. У дослідженнях було дозволено застосування гастропротекторних препаратів і ацетилсаліцилової кислоти у низьких дозах.
Загальна безпека
Не було суттєвих відмінностей у частоті тромботичних серцево-судинних ускладнень при застосуванні еторикоксибу і диклофенаку. Кардіоренальні побічні реакції частіше спостерігалися при застосуванні еторикоксибу, ніж диклофенаку; цей ефект був дозозалежним (детально про результати див. нижче). Побічні реакції з боку ШКТ і печінки виникали значно частіше при застосуванні диклофенаку, ніж еторикоксибу. Частота виникнення побічних реакцій в дослідженнях EDGE і EDGE II, а також побічних реакцій, що розглядалися як серйозні або такі, що призводять до відміни препарату в дослідженні MEDAL, була вищою при застосуванні еторикоксибу, ніж диклофенаку.
Безпека відносно серцево-судинної системи
Частота підтверджених тромботичних серцево-судинних серйозних побічних реакцій (включаючи реакції з боку серця, цереброваскулярні реакції і реакції з боку периферичних судин) була порівнянною у еторикоксибу і диклофенаку (дані підсумовано в таблиці 1). Не було суттєвих відмінностей в показниках частоти тромботичних ускладнень при застосуванні еторикоксибу і диклофенаку в усіх проаналізованих підгрупах, включаючи пацієнтів із кардіоваскулярним ризиком. При окремому розгляді відносний ризик виникнення підтверджених серйозних тромботичних побічних реакцій з боку серцево-судинної системи при застосуванні еторикоксибу в дозі 60 мг або 90 мг і диклофенаку в дозі 150 мг був однаковим.
Таблиця 1
Показники підтверджених тромботичних ускладнень з боку серцево-судинної системи (об’єднана програма MEDAL)
|
Ускладнення |
Еторикоксиб (N=16819) 25836 пацієнто-років |
Диклофенак (N=16483) 24766 пацієнто-років |
Порівняння між групами лікування |
|||
|
Показник† (95 % ДІ) |
Показник† (95 % ДІ) |
Відносний ризик (95 % ДІ) |
||||
|
Підтверджені серйозні тромботичні побічні реакції з боку серцево-судинної системи |
||||||
|
За протоколом |
1,24 (1,11; 1,38) |
1,30 (1,17; 1,45) |
0,95 (0,81; 1,11) |
|||
|
За наміром лікуватися |
1,25 (1,14; 1,36) |
1,19 (1,08; 1,30) |
1,05 (0,93; 1,19) |
|||
|
Підтверджені ускладнення з боку серця |
||||||
|
За протоколом |
0,71 (0,61; 0,82) |
0,78 (0,68; 0,90) |
0,90 (0,74; 1,10) |
|||
|
За наміром лікуватися |
0,69 (0,61; 0,78) |
0,70 (0,62; 0,79) |
0,99 (0,84; 1,17) |
|||
|
Підтверджені цереброваскулярні ускладнення |
||||||
|
За протоколом |
0,34 (0,28; 0,42) |
0,32 (0,25; 0,40) |
1,08 (0,80; 1,46) |
|||
|
За наміром лікуватися |
0,33 (0,28; 0,39) |
0,29 (0,24; 0,35) |
1,12 (0,87; 1,44) |
|||
|
Підтверджені ускладнення з боку периферичних судин |
||||||
|
За протоколом |
0,20 (0,15; 0,27) |
0,22 (0,17; 0,29) |
0,92 (0,63; 1,35) |
|||
|
За наміром лікуватися |
0,24 (0,20; 0,30) |
0,23 (0,18; 0,28) |
1,08 (0,81; 1,44) |
|||
†Ускладнень на 100 пацієнто-років; ДІ – довірчий інтервал.
N – загальна кількість пацієнтів у популяції за протоколом.
За протоколом: усі ускладнення під час досліджуваної терапії або протягом 14 днів після її припинення (за винятком пацієнтів, які прийняли <75 % досліджуваного препарату або приймали недосліджувані нестероїдні протизапальні препарати >10 % всього періоду).
За наміром лікуватися: всі підтверджені ускладнення до закінчення дослідження (у т. ч. у пацієнтів, які могли зазнати втручання, не пов’язаного з дослідженням, з подальшим припиненням прийому досліджуваного препарату). Загальна кількість рандомізованих пацієнтів: 17412 у групі еторикоксибу і 17289 у групі диклофенаку.
Показник серцево-судинної смертності, як і загальної смертності, був подібним у групах лікування еторикоксибом і диклофенаком.
Кардіоренальні ускладнення
Приблизно 50 % пацієнтів, прийнятих у дослідження MEDAL, мали артеріальну гіпертензію в анамнезі на початковому етапі. У цьому дослідженні частота припинення лікування унаслідок виникнення побічних реакцій, пов’язаних із артеріальною гіпертензією, була статистично значно вищою у групі застосування еторикоксибу, ніж у групі диклофенаку. Частота такої побічної реакції, як застійна серцева недостатність (припинення прийому препарату і серйозні реакції), була аналогічною як при прийомі еторикоксибу 60 мг, так і при прийомі диклофенаку 150 мг, проте частота виникнення цих реакцій була вищою при прийомі еторикоксибу 90 мг порівняно з диклофенаком 150 мг (статистично значуща різниця при прийомі еторикоксибу 90 мг порівняно з 150 мг диклофенаку в групі ОА MEDAL). Частота підтверджених побічних реакцій, пов’язаних із застійною серцевою недостатністю (явища, що були серйозними і вимагали госпіталізації або невідкладної допомоги), була незначно вищою при прийомі еторикоксибу порівняно з прийомом диклофенаку 150 мг, і цей ефект залежав від дози. Частота припинення лікування унаслідок виникнення побічних реакцій, пов’язаних з набряками, була значно вищою при прийомі еторикоксибу порівняно з прийомом диклофенаку 150 мг, і цей ефект залежав від дози (статистично значуща різниця при прийомі еторикоксибу 90 мг, але не еторикоксибу 60 мг).
Кардіоренальні результати, отримані в дослідженнях EDGE і EDGE II, відповідали даним, про які повідомлялося в дослідженні MEDAL.
В окремих дослідженнях програми MEDAL абсолютна частота припинення лікування в будь-якій групі лікування еторикоксибом (60 мг або 90 мг) становила до 2,6 % при артеріальній гіпертензії, до 1,9 % при набряках і до 1,1 % при застійній серцевій недостатності, при цьому вища частота відміни препарату спостерігалася у разі прийому еторикоксибу 90 мг, ніж 60 мг.
Результати шлунково-кишкової переносимості в програмі MEDAL
Значно менший показник відміни препарату внаслідок виникнення будь-якого клінічного ускладнення з боку ШКТ (наприклад диспепсії, абдомінального болю, виразки) спостерігався при застосуванні еторикоксибу, ніж диклофенаку, в кожному з трьох досліджень програми MEDAL. Показники відміни препарату внаслідок клінічних реакцій з боку ШКТ на 100 пацієнто‑років за весь період дослідження були такими: 3,23 для еторикоксибу і 4,96 для диклофенаку в дослідженні MEDAL; 9,12 для еторикоксибу і 12,28 для диклофенаку в дослідженні EDGE; 3,71 для еторикоксибу і 4,81 для диклофенаку в дослідженні EDGE II.
Результати програми MEDAL щодо безпеки для ШКТ
Загальні реакції з боку верхнього відділу ШКТ були визначені як перфорації, виразки і кровотечі. Підгрупа загальних реакцій з боку верхнього відділу ШКТ, які вважалися ускладненими, включала перфорації, обструкції і ускладнені кровотечі; підгрупа загальних реакцій з боку верхнього відділу ШКТ, які вважалися неускладненими, включала неускладнені кровотечі і неускладнені виразки. Значно менший показник частоти загальних реакцій з боку верхніх відділів ШКТ спостерігався при застосуванні еторикоксибу, ніж диклофенаку. Не було суттєвої різниці між еторикоксибом і диклофенаком щодо показника частоти ускладнених реакцій. Для підгрупи таких реакцій, як кровотеча у верхньому відділі ШКТ (об’єднані ускладнені і неускладнені), не було суттєвої відмінності між еторикоксибом і диклофенаком. Перевага еторикоксибу щодо впливу на верхній відділ ШКТ порівняно з диклофенаком не була статистично значущою у пацієнтів, що одночасно застосовують аспірин в низьких дозах (приблизно 33 % пацієнтів).
Показник частоти на 100 пацієнто-років підтверджених ускладнених і неускладнених клінічних реакцій з боку верхнього відділу ШКТ (перфорації, виразки і кровотечі) становив 0,67 (95 % ДІ 0,57, 0,77) при прийомі еторикоксибу і 0,97 (95 % ДІ 0,85, 1,10) при прийомі диклофенаку, при цьому відносний ризик становив 0,69 (95 % ДІ 0,57, 0,83).
Визначався показник частоти підтверджених реакцій з боку верхнього відділу ШКТ у пацієнтів літнього віку; найбільше зниження спостерігалося у пацієнтів віком ≥75 років (1,35 [95 % ДІ 0,94, 1,87] реакцій на 100 пацієнтів-років при прийомі еторикоксибу порівняно з 2,78 [95 % ДІ 2,14, 3,56] при прийомі диклофенаку).
Показники частоти підтверджених клінічних реакцій з боку нижнього відділу ШКТ (перфорація тонкого або товстого кишечнику, обструкція або кровотеча) статистично не відрізнялися при застосуванні еторикоксибу і диклофенаку.
Результати програми MEDAL щодо безпеки для печінки
Еторикоксиб був асоційований із статистично значно меншою частотою відміни препарату унаслідок виникнення побічних реакцій з боку печінки, ніж диклофенак. У об’єднаній програмі MEDAL 0,3 % пацієнтів, що застосовували еторикоксиб, і 2,7 % пацієнтів, що застосовували диклофенак, припинили застосування препарату внаслідок виникнення побічних реакцій з боку печінки. Показник на 100 пацієнто-років становив 0,22 при застосуванні еторикоксибу і 1,84 при застосуванні диклофенаку (р-значення було < 0,001 для еторикоксибу порівняно з диклофенаком). Проте в програмі MEDAL більшість побічних реакцій з боку печінки були несерйозними.
Додаткові дані з безпеки для серцево-судинної системи щодо тромботичних ускладнень
У ході клінічних досліджень, за винятком досліджень програми MEDAL, приблизно 3100 пацієнтів отримували еторикоксиб у дозах ≥ 60 мг на добу упродовж 12 тижнів і довше. Не було значущої відмінності показників підтверджених серйозних тромботичних серцево-судинних ускладнень у пацієнтів, які приймали еторикоксиб у дозі ≥ 60 мг, плацебо або інші НПЗП (за винятком напроксену). Проте частота таких реакцій була вищою у пацієнтів, які отримували еторикоксиб, порівняно з тими, хто отримував напроксен у дозі 500 мг 2 рази на добу. Відмінність антитромботичної активності між деякими НПЗП, що інгібують ЦОГ-1, і селективними інгібіторами ЦОГ-2 може бути клінічно значущою у пацієнтів групи ризику виникнення тромбоемболічних ускладнень. Селективні інгібітори ЦОГ-2 знижують утворення системного (і тому, можливо, ендотеліального) простацикліну без впливу на тромбоцитарний тромбоксан. Клінічне значення цих даних невідоме.
Додаткові дані щодо безпеки для ШКТ
Під час двох 12-тижневих подвійно сліпих ендоскопічних досліджень кумулятивна частота виникнення гастродуоденальних виразок була значно нижчою у пацієнтів, які отримували лікування еторикоксибом у дозі 120 мг 1 раз на добу, ніж у пацієнтів, які отримували лікування напроксеном у дозі 500 мг 2 рази на добу або ібупрофеном у дозі 800 мг 3 рази на добу. Частота виникнення виразок була вищою при застосуванні еторикоксибу, ніж плацебо.
Дослідження функції нирок у пацієнтів літнього віку
У ході рандомізованого, подвійно сліпого, плацебо-контрольованого дослідження з паралельними групами оцінювався вплив 15-денного лікування еторикоксибом (90 мг), целекоксибом (200 мг 2 рази на добу), напроксеном (500 мг 2 рази на добу) і плацебо на виведення натрію з сечею, артеріальний тиск та інші показники функції нирок у пацієнтів від 60 до 85 років, що дотримуються дієти із вмістом солі 200 мЕкв/добу. Еторикоксиб, целекоксиб і напроксен мали подібний вплив на виведення натрію з сечею при 2-тижневому лікуванні. Усі активні препарати порівняння показали підвищення відносно плацебо систолічного артеріального тиску; проте еторикоксиб асоціювався зі статистично значущим підвищенням на 14-й день порівняно з целекоксибом і напроксеном (середня зміна систолічного тиску порівняно з початковим рівнем
Атрибути
| Торгова назва | Organon Central East Gmbh |
| Країна виробництва | Швейцарія 2528 |
| Умови продажу | Рецепт |
| Діючі речовини | Еторикоксиб |
| АТХ-група | M01A H05 |
| Медикаменти | Нестероїдні протизапальні препарати |
Інструкція
Аркоксія (arcoxia) інструкція по застосуванню
Особливості застосування препарату
Спосіб застосування та дози Аркоксія
Склад
діюча речовина: etoricoxib;
- 1 таблетка містить 30 мг, 60 мг, 90 мг або 120 мг еторикоксибу;
допоміжні речовини: кальцію гідрофосфат безводний, целюлоза мікрокристалічна, натрію кроскармелоза, магнію стеарат;
оболонка таблетки: барвник Опадрай II синьо-зелений 39К11526 (для дозування 30 мг), Опадрай II зелений 39К11520 (для дозування 60 мг), Опадрай II білий 39К18305 (для дозування 90 мг), Опадрай II зелений 39К11529 (для дозування 120 мг), віск карнаубський;
склад барвника Опадрай II синьо-зелений 39К11526, Опадрай II зелений 39К11520, Опадрай II зелений 39К11529: лактоза, моногідрат; гіпромелоза; титану діоксид (Е 171); триацетин; індигокармін (Е 132); заліза оксид жовтий (Е 172);
склад барвника Опадрай II білий 39К18305: лактоза, моногідрат; гіпромелоза; титану діоксид (Е 171); триацетин.
Лікарська форма
Таблетки, вкриті плівковою оболонкою.
Основні фізико-хімічні властивості:
- Таблетки по 30 мг: двоопуклі таблетки яблукоподібної форми, вкриті плівковою оболонкою синьо-зеленого кольору, з гравіруванням <101> з одного боку та <ACX 30> з іншого.
- Таблетки по 60 мг: двоопуклі таблетки яблукоподібної форми, вкриті плівковою оболонкою темно-зеленого кольору, з гравіруванням <200> з одного боку та <ARCOXIA 60> з іншого.
- Таблетки по 90 мг: двоопуклі таблетки яблукоподібної форми, вкриті плівковою оболонкою білого кольору, з гравіруванням <202> з одного боку та <ARCOXIA 90> з іншого.
- Таблетки по 120 мг: двоопуклі таблетки яблукоподібної форми, вкриті плівковою оболонкою блідо-зеленого кольору, з гравіруванням <204> з одного боку та <ARCOXIA 120> з іншого.
Фармако-терапевтична група
Нестероїдні протизапальні та протиревматичні препарати. Коксиби. Код АТХ M01A H05.
Фармакологічні властивості
Фармакодинаміка.
Механізм дії
Еторикоксиб є пероральним селективним інгібітором циклооксигенази-2 (ЦОГ-2) у межах клінічного діапазону доз.
У ході клінічних фармакологічних досліджень лікарський засіб Аркоксія дозозалежно інгібував ЦОГ-2 без інгібування ЦОГ-1 при застосуванні у дозах до 150 мг на добу. Еторикоксиб не інгібує синтез простагландинів шлунка та не впливає на функцію тромбоцитів.
Циклооксигеназа відповідає за утворення простагландинів. Ідентифіковано дві ізоформи – ЦОГ-1 та ЦОГ-2. ЦОГ-2 є ізоформою ферменту, що індукується імпульсом прозапалення та розглядається як основний фактор, що відповідає за синтез простаноїдних медіаторів болю, запалення та гарячки. ЦОГ-2 також задіяна у процесах овуляції, імплантації та закриття артеріальної протоки, регуляції функції нирок та центральної нервової системи (індукція гарячки, відчуття болю, когнітивна функція). Також може брати участь у процесі загоєння виразок. ЦОГ-2 було ідентифіковано у тканині навколо виразки шлунка у людини, але значення для загоєння виразки не встановлено.
Ефективність
У пацієнтів з остеоартритом еторикоксиб у дозі 60 мг 1 раз на добу значно покращує стан при болях та оцінку пацієнта щодо стану захворювання. Ці позитивні ефекти спостерігалися вже на другий день лікування і зберігалися протягом періоду до 52 тижнів. У ході досліджень із застосуванням еторикоксибу в дозі 30 мг 1 раз на добу ефективність цього препарату перевищувала плацебо протягом 12 тижнів лікування (використовувалися оцінки, що застосовувалися в інших дослідженнях). Під час дослідження підбору дози еторикоксиб у дозі 60 мг демонстрував значно більш виражене покращення, ніж в дозі 30 мг, відносно усіх 3 основних кінцевих точок після 6 тижнів лікування. Застосування дози 30 мг при остеоартриті кисті не вивчалося.
У пацієнтів з ревматоїдним артритом еторикоксиб у дозі 60 мг та 90 мг 1 раз на добу значно покращував стан стосовно вираженості болю, запалення, а також рухливості. У ході досліджень з оцінки доз 60 мг та 90 мг позитивні ефекти зберігалися протягом 12-тижневого періоду лікування. У ході дослідження з оцінки дози 60 мг порівняно з дозою 90 мг, обидва дозування еторикоксибу – 60 мг 1 раз на добу та 90 мг 1 раз на добу – були більш ефективні ніж плацебо. Доза 90 мг була ефективніша, ніж доза 60 мг відповідно до методу Загальної оцінки болю пацієнтів (0–100 мм візуальна аналогова шкала), з середнім покращенням -2,71 мм (95 % ДІ:
-4,98 мм, -0,45 мм).
У пацієнтів з нападами гострого подагричного артриту еторикоксиб у дозі 120 мг 1 раз на добу протягом 8 днів полегшував біль у суглобах середнього та тяжкого ступеня та запалення порівняно з індометацином у дозі 50 мг 3 рази на добу. Зменшення вираженості болю спостерігається вже через 4 години після початку лікування.
У пацієнтів із анкілозуючим спондилітом еторикоксиб у дозі 90 мг 1 раз на добу забезпечує значне покращення при болю у хребті, запаленні, обмеженні рухів, а також покращує функціональну здатність. Клінічні переваги еторикоксибу спостерігалися на другий день після початку терапії і зберігалися упродовж 52-тижневого періоду лікування. У ході другого дослідження з оцінки дози 60 мг порівняно з дозою 90 мг, еторикоксиб у дозі 60 мг 1 раз на добу та 90 мг 1 раз на добу продемонстрував схожу ефективність порівняно з напроксеном 1000 мг щоденно. У пацієнтів, що не демонстрували адекватної відповіді під час застосування дози 60 мг щоденно протягом 6 тижнів, підвищення дози до 90 мг щоденно покращувало оцінку інтенсивності болю у спині (0–100 мм візуальна аналогова шкала) порівняно з продовженням прийому 60 мг щоденно, з середнім покращенням -2,70 мм (95 % ДІ: -4,88 мм, -0,52 мм).
Під час клінічного дослідження післяопераційного зубного болю еторикоксиб у дозі 90 мг застосовувався 1 раз на добу до трьох днів. У підгрупі пацієнтів з помірним болем в початковому стані еторикоксиб у дозі 90 мг демонстрував знеболюючий ефект, подібний до такого в ібупрофену 600 мг (16,11 проти 16,39; P=0,722), і перевищував ефект парацетамолу/кодеїну 600 мг/60 мг (11,00; P<0,001) і плацебо (6,84; P<0,001), що визначалося за показником повного полегшення болю через 6 годин (TOPAR6). Кількість пацієнтів, які повідомляли про застосування препаратів екстреного знеболення протягом 24 годин, склала 40,8 % у групі застосування еторикоксибу 90 мг, 25,5 % у групі застосування ібупрофену 600 мг кожні 6 годин і 46,7 % у групі застосування парацетамолу/кодеїну 600 мг/60 мг кожні 6 годин порівняно з 76,2 % пацієнтів, які приймали плацебо. У цьому дослідженні початок аналгезивної дії (відчутне полегшення болю) 90 мг еторикоксибу спостерігалося вже через 28 хвилин після прийому препарату.
Безпека
Міжнародна дослідницька програма тривалого застосування еторикоксибу і диклофенаку при артриті (MEDAL)
Програма MEDAL була проспективно розробленою програмою відносно результатів щодо безпеки з боку серцево-судинної системи, отриманих за об’єднаними даними трьох рандомізованих, подвійно сліпих, контрольованих активним препаратом порівняння досліджень (дослідження MEDAL, EDGE II і EDGE).
У дослідженні MEDAL, що було направлене на визначення впливу на серцево-судинну систему, брали участь 17804 пацієнти з остеоартритом (ОА) і 5700 ‒ з ревматоїдним артритом (РА), які застосовували еторикоксиб у дозі 60 мг (ОА) чи 90 мг (ОА і РА) або диклофенак у дозі 150 мг на добу протягом в середньому 20,3 місяця (максимально ‒ 42,3 місяця, медіана ‒ 21,3 місяця). У цьому дослідженні були зафіксовані тільки серйозні побічні реакції і припинення прийому препарату внаслідок виникнення будь-яких побічних реакцій.
У ході досліджень EDGE і EDGE II порівнювали шлунково-кишкову переносимість еторикоксибу і диклофенаку. У дослідженні EDGE брали участь 7111 пацієнтів з ОА, які отримували еторикоксиб у дозі 90 мг на добу (у 1,5 раза вище рекомендованої дози для лікування ОА), або диклофенак у дозі 150 мг на добу протягом в середньому 9,1 місяця (максимум ‒ 16,6 місяця, медіана ‒ 11,4 місяця). У дослідженні EDGE II брали участь 4086 пацієнтів з РА, які отримували лікування еторикоксибом у дозі 90 мг на добу або диклофенаком у дозі 150 мг на добу протягом в середньому 19,2 місяця (максимум ‒ 33,1 місяця, медіана ‒ 24 місяці).
У об’єднаній програмі MEDAL брали участь 34701 пацієнт з ОА і РА, які отримували лікування упродовж періоду в середньому 17,9 місяця (максимум ‒ 42,3 місяця, медіана ‒ 16,3 місяця); приблизно 12800 пацієнтів отримували лікування понад 24 місяці. У пацієнтів, зареєстрованих у цій програмі, були різні початкові чинники ризику стосовно серцево-судинної системи і шлунково-кишкового тракту (ШКТ). Пацієнти з нещодавно перенесеним інфарктом міокарда, аортокоронарним шунтуванням або черезшкірною коронарною ангіопластикою упродовж 6 місяців до реєстрації в дослідженні були виключені з дослідження. У дослідженнях було дозволено застосування гастропротекторних препаратів і ацетилсаліцилової кислоти у низьких дозах.
Загальна безпека
Не було суттєвих відмінностей у частоті тромботичних серцево-судинних ускладнень при застосуванні еторикоксибу і диклофенаку. Кардіоренальні побічні реакції частіше спостерігалися при застосуванні еторикоксибу, ніж диклофенаку; цей ефект був дозозалежним (детально про результати див. нижче). Побічні реакції з боку ШКТ і печінки виникали значно частіше при застосуванні диклофенаку, ніж еторикоксибу. Частота виникнення побічних реакцій в дослідженнях EDGE і EDGE II, а також побічних реакцій, що розглядалися як серйозні або такі, що призводять до відміни препарату в дослідженні MEDAL, була вищою при застосуванні еторикоксибу, ніж диклофенаку.
Безпека відносно серцево-судинної системи
Частота підтверджених тромботичних серцево-судинних серйозних побічних реакцій (включаючи реакції з боку серця, цереброваскулярні реакції і реакції з боку периферичних судин) була порівнянною у еторикоксибу і диклофенаку (дані підсумовано в таблиці 1). Не було суттєвих відмінностей в показниках частоти тромботичних ускладнень при застосуванні еторикоксибу і диклофенаку в усіх проаналізованих підгрупах, включаючи пацієнтів із кардіоваскулярним ризиком. При окремому розгляді відносний ризик виникнення підтверджених серйозних тромботичних побічних реакцій з боку серцево-судинної системи при застосуванні еторикоксибу в дозі 60 мг або 90 мг і диклофенаку в дозі 150 мг був однаковим.
Таблиця 1
Показники підтверджених тромботичних ускладнень з боку серцево-судинної системи (об’єднана програма MEDAL)
|
Ускладнення |
Еторикоксиб (N=16819) 25836 пацієнто-років |
Диклофенак (N=16483) 24766 пацієнто-років |
Порівняння між групами лікування |
|||
|
Показник† (95 % ДІ) |
Показник† (95 % ДІ) |
Відносний ризик (95 % ДІ) |
||||
|
Підтверджені серйозні тромботичні побічні реакції з боку серцево-судинної системи |
||||||
|
За протоколом |
1,24 (1,11; 1,38) |
1,30 (1,17; 1,45) |
0,95 (0,81; 1,11) |
|||
|
За наміром лікуватися |
1,25 (1,14; 1,36) |
1,19 (1,08; 1,30) |
1,05 (0,93; 1,19) |
|||
|
Підтверджені ускладнення з боку серця |
||||||
|
За протоколом |
0,71 (0,61; 0,82) |
0,78 (0,68; 0,90) |
0,90 (0,74; 1,10) |
|||
|
За наміром лікуватися |
0,69 (0,61; 0,78) |
0,70 (0,62; 0,79) |
0,99 (0,84; 1,17) |
|||
|
Підтверджені цереброваскулярні ускладнення |
||||||
|
За протоколом |
0,34 (0,28; 0,42) |
0,32 (0,25; 0,40) |
1,08 (0,80; 1,46) |
|||
|
За наміром лікуватися |
0,33 (0,28; 0,39) |
0,29 (0,24; 0,35) |
1,12 (0,87; 1,44) |
|||
|
Підтверджені ускладнення з боку периферичних судин |
||||||
|
За протоколом |
0,20 (0,15; 0,27) |
0,22 (0,17; 0,29) |
0,92 (0,63; 1,35) |
|||
|
За наміром лікуватися |
0,24 (0,20; 0,30) |
0,23 (0,18; 0,28) |
1,08 (0,81; 1,44) |
|||
†Ускладнень на 100 пацієнто-років; ДІ – довірчий інтервал.
N – загальна кількість пацієнтів у популяції за протоколом.
За протоколом: усі ускладнення під час досліджуваної терапії або протягом 14 днів після її припинення (за винятком пацієнтів, які прийняли <75 % досліджуваного препарату або приймали недосліджувані нестероїдні протизапальні препарати >10 % всього періоду).
За наміром лікуватися: всі підтверджені ускладнення до закінчення дослідження (у т. ч. у пацієнтів, які могли зазнати втручання, не пов’язаного з дослідженням, з подальшим припиненням прийому досліджуваного препарату). Загальна кількість рандомізованих пацієнтів: 17412 у групі еторикоксибу і 17289 у групі диклофенаку.
Показник серцево-судинної смертності, як і загальної смертності, був подібним у групах лікування еторикоксибом і диклофенаком.
Кардіоренальні ускладнення
Приблизно 50 % пацієнтів, прийнятих у дослідження MEDAL, мали артеріальну гіпертензію в анамнезі на початковому етапі. У цьому дослідженні частота припинення лікування унаслідок виникнення побічних реакцій, пов’язаних із артеріальною гіпертензією, була статистично значно вищою у групі застосування еторикоксибу, ніж у групі диклофенаку. Частота такої побічної реакції, як застійна серцева недостатність (припинення прийому препарату і серйозні реакції), була аналогічною як при прийомі еторикоксибу 60 мг, так і при прийомі диклофенаку 150 мг, проте частота виникнення цих реакцій була вищою при прийомі еторикоксибу 90 мг порівняно з диклофенаком 150 мг (статистично значуща різниця при прийомі еторикоксибу 90 мг порівняно з 150 мг диклофенаку в групі ОА MEDAL). Частота підтверджених побічних реакцій, пов’язаних із застійною серцевою недостатністю (явища, що були серйозними і вимагали госпіталізації або невідкладної допомоги), була незначно вищою при прийомі еторикоксибу порівняно з прийомом диклофенаку 150 мг, і цей ефект залежав від дози. Частота припинення лікування унаслідок виникнення побічних реакцій, пов’язаних з набряками, була значно вищою при прийомі еторикоксибу порівняно з прийомом диклофенаку 150 мг, і цей ефект залежав від дози (статистично значуща різниця при прийомі еторикоксибу 90 мг, але не еторикоксибу 60 мг).
Кардіоренальні результати, отримані в дослідженнях EDGE і EDGE II, відповідали даним, про які повідомлялося в дослідженні MEDAL.
В окремих дослідженнях програми MEDAL абсолютна частота припинення лікування в будь-якій групі лікування еторикоксибом (60 мг або 90 мг) становила до 2,6 % при артеріальній гіпертензії, до 1,9 % при набряках і до 1,1 % при застійній серцевій недостатності, при цьому вища частота відміни препарату спостерігалася у разі прийому еторикоксибу 90 мг, ніж 60 мг.
Результати шлунково-кишкової переносимості в програмі MEDAL
Значно менший показник відміни препарату внаслідок виникнення будь-якого клінічного ускладнення з боку ШКТ (наприклад диспепсії, абдомінального болю, виразки) спостерігався при застосуванні еторикоксибу, ніж диклофенаку, в кожному з трьох досліджень програми MEDAL. Показники відміни препарату внаслідок клінічних реакцій з боку ШКТ на 100 пацієнто‑років за весь період дослідження були такими: 3,23 для еторикоксибу і 4,96 для диклофенаку в дослідженні MEDAL; 9,12 для еторикоксибу і 12,28 для диклофенаку в дослідженні EDGE; 3,71 для еторикоксибу і 4,81 для диклофенаку в дослідженні EDGE II.
Результати програми MEDAL щодо безпеки для ШКТ
Загальні реакції з боку верхнього відділу ШКТ були визначені як перфорації, виразки і кровотечі. Підгрупа загальних реакцій з боку верхнього відділу ШКТ, які вважалися ускладненими, включала перфорації, обструкції і ускладнені кровотечі; підгрупа загальних реакцій з боку верхнього відділу ШКТ, які вважалися неускладненими, включала неускладнені кровотечі і неускладнені виразки. Значно менший показник частоти загальних реакцій з боку верхніх відділів ШКТ спостерігався при застосуванні еторикоксибу, ніж диклофенаку. Не було суттєвої різниці між еторикоксибом і диклофенаком щодо показника частоти ускладнених реакцій. Для підгрупи таких реакцій, як кровотеча у верхньому відділі ШКТ (об’єднані ускладнені і неускладнені), не було суттєвої відмінності між еторикоксибом і диклофенаком. Перевага еторикоксибу щодо впливу на верхній відділ ШКТ порівняно з диклофенаком не була статистично значущою у пацієнтів, що одночасно застосовують аспірин в низьких дозах (приблизно 33 % пацієнтів).
Показник частоти на 100 пацієнто-років підтверджених ускладнених і неускладнених клінічних реакцій з боку верхнього відділу ШКТ (перфорації, виразки і кровотечі) становив 0,67 (95 % ДІ 0,57, 0,77) при прийомі еторикоксибу і 0,97 (95 % ДІ 0,85, 1,10) при прийомі диклофенаку, при цьому відносний ризик становив 0,69 (95 % ДІ 0,57, 0,83).
Визначався показник частоти підтверджених реакцій з боку верхнього відділу ШКТ у пацієнтів літнього віку; найбільше зниження спостерігалося у пацієнтів віком ≥75 років (1,35 [95 % ДІ 0,94, 1,87] реакцій на 100 пацієнтів-років при прийомі еторикоксибу порівняно з 2,78 [95 % ДІ 2,14, 3,56] при прийомі диклофенаку).
Показники частоти підтверджених клінічних реакцій з боку нижнього відділу ШКТ (перфорація тонкого або товстого кишечнику, обструкція або кровотеча) статистично не відрізнялися при застосуванні еторикоксибу і диклофенаку.
Результати програми MEDAL щодо безпеки для печінки
Еторикоксиб був асоційований із статистично значно меншою частотою відміни препарату унаслідок виникнення побічних реакцій з боку печінки, ніж диклофенак. У об’єднаній програмі MEDAL 0,3 % пацієнтів, що застосовували еторикоксиб, і 2,7 % пацієнтів, що застосовували диклофенак, припинили застосування препарату внаслідок виникнення побічних реакцій з боку печінки. Показник на 100 пацієнто-років становив 0,22 при застосуванні еторикоксибу і 1,84 при застосуванні диклофенаку (р-значення було < 0,001 для еторикоксибу порівняно з диклофенаком). Проте в програмі MEDAL більшість побічних реакцій з боку печінки були несерйозними.
Додаткові дані з безпеки для серцево-судинної системи щодо тромботичних ускладнень
У ході клінічних досліджень, за винятком досліджень програми MEDAL, приблизно 3100 пацієнтів отримували еторикоксиб у дозах ≥ 60 мг на добу упродовж 12 тижнів і довше. Не було значущої відмінності показників підтверджених серйозних тромботичних серцево-судинних ускладнень у пацієнтів, які приймали еторикоксиб у дозі ≥ 60 мг, плацебо або інші НПЗП (за винятком напроксену). Проте частота таких реакцій була вищою у пацієнтів, які отримували еторикоксиб, порівняно з тими, хто отримував напроксен у дозі 500 мг 2 рази на добу. Відмінність антитромботичної активності між деякими НПЗП, що інгібують ЦОГ-1, і селективними інгібіторами ЦОГ-2 може бути клінічно значущою у пацієнтів групи ризику виникнення тромбоемболічних ускладнень. Селективні інгібітори ЦОГ-2 знижують утворення системного (і тому, можливо, ендотеліального) простацикліну без впливу на тромбоцитарний тромбоксан. Клінічне значення цих даних невідоме.
Додаткові дані щодо безпеки для ШКТ
Під час двох 12-тижневих подвійно сліпих ендоскопічних досліджень кумулятивна частота виникнення гастродуоденальних виразок була значно нижчою у пацієнтів, які отримували лікування еторикоксибом у дозі 120 мг 1 раз на добу, ніж у пацієнтів, які отримували лікування напроксеном у дозі 500 мг 2 рази на добу або ібупрофеном у дозі 800 мг 3 рази на добу. Частота виникнення виразок була вищою при застосуванні еторикоксибу, ніж плацебо.
Дослідження функції нирок у пацієнтів літнього віку
У ході рандомізованого, подвійно сліпого, плацебо-контрольованого дослідження з паралельними групами оцінювався вплив 15-денного лікування еторикоксибом (90 мг), целекоксибом (200 мг 2 рази на добу), напроксеном (500 мг 2 рази на добу) і плацебо на виведення натрію з сечею, артеріальний тиск та інші показники функції нирок у пацієнтів від 60 до 85 років, що дотримуються дієти із вмістом солі 200 мЕкв/добу. Еторикоксиб, целекоксиб і напроксен мали подібний вплив на виведення натрію з сечею при 2-тижневому лікуванні. Усі активні препарати порівняння показали підвищення відносно плацебо систолічного артеріального тиску; проте еторикоксиб асоціювався зі статистично значущим підвищенням на 14-й день порівняно з целекоксибом і напроксеном (середня зміна систолічного тиску порівняно з початковим рівнем
Завантаження
Підтвердьте свою дію
Ви дійсно бажаєте виконати цю дію?
Цей товар було додано в кошик